【编者按】
每年2月最后一天是“国际罕见病日”,2023年2月28日,也是第十六个“国际罕见病日”。
罕见病又称“孤儿病”,指患病率很低、很少见的疾病。目前确定的罕见病病种约7000种,影响全球3亿人次。虽然每一种罕见病的患者不多,累加起来的总和发病率却达到1-1.5%。也就是说,每100名新生儿中,就会有至少1名是罕见病患儿。
中国的罕见病总患病人口数接近2000万,还有数倍的基因携带者。因此,罕见病在中国并不罕见。并且,罕见病患者中,大部分都涉及遗传病,其中还有许多人没有意识到疾病就发生在自己身上。
“国际罕见病日”到来之际,澎湃新闻通过口述形式,记录罕见病患者的生活境遇。希望通过这些真实的故事、经历,唤起社会大众对这一群体的关注,给予他们更多帮助。
1.73米的个子,43码的鞋子,笑笑(化名)本以为自己只是生下来长得像妈妈,手长脚长,直到2015年她的左眼突然发生晶体脱位,差点失明,她才知道,自己患了一种罕见病:马凡综合征。
马凡综合征,也称为马方综合征,这一疾病已被纳入到中国第一批罕见病目录中。
有人将这种疾病称为“天才病”,它较为常见的临床表现包括四肢和手指较长、趾细长不匀称、身高明显超出常人等。公开资料显示,著名小提琴家帕格尼尼、美国女排名将海曼、美国领导人林肯都罹患此病。
罕见病也让笑笑饱受煎熬。伴随着左眼晶体脱位而来的,是血管瘤。一次次奔波在大医院,眼科手术、心脏手术……她的成长经历中,还失去了两位最亲的人:母亲和哥哥。
笑笑11岁那年,母亲在饭后说了句不舒服想躺一会儿,睡着睡着就没有了呼吸;哥哥一次胸口痛后被诊断为风湿性心脏病,因无钱换心脏治疗,26岁就走了。医生推断,笑笑两位亲人离世,或都与马凡综合征有关。
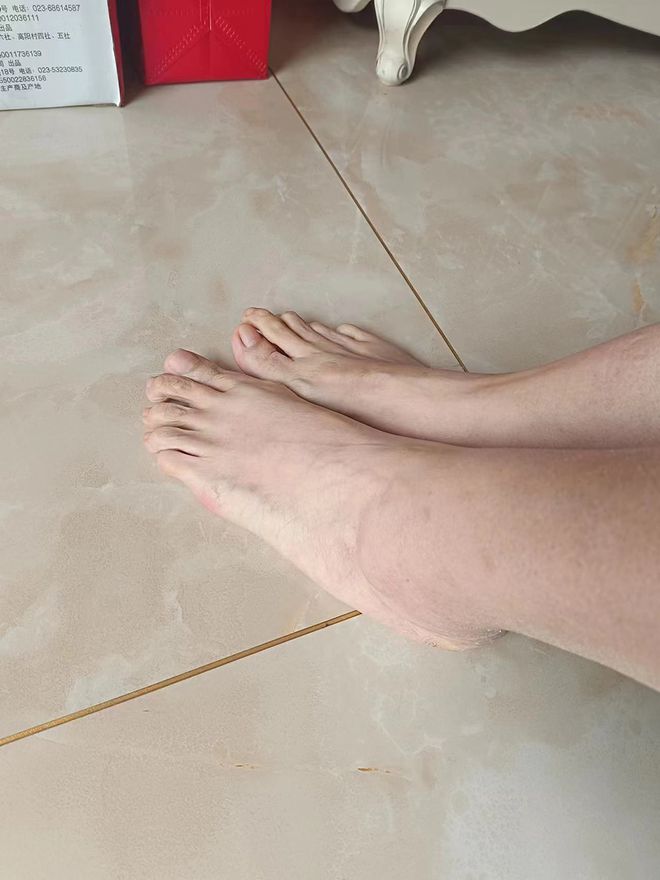
笑笑的脚,较常人的脚大出许多,有43码。本文图片均为 受访者 供图

以下是笑笑的自述:
手长脚长,可能是一种病?
我今年42岁,来自四川凉山州一个农村家庭。回想起我小时候的日子,爸爸、妈妈、哥哥和我,一家四口,生活原本平淡美好,但不幸却一次次袭来。
我的家庭变故从我11岁那个夜里开始。那晚,妈妈说她不舒服想去躺一会儿,就进了卧室。爸爸忙完了手上的活,叫了一声妈妈,没有得到她的回应,进入睡房一看,妈妈横躺在床上,爸爸开玩笑地掐了下妈妈的腿说道:你要睡觉就好好躺着睡。妈妈还是没有应答,爸爸着急了,用手凑到了妈妈的鼻子下,她已经没有呼吸了。
妈妈的意外离去,让我们这个本就贫困的家庭更为捉襟见肘。为了节省开支,读六年级的我辍学了,跟着爸爸下地干起了农活。为了家庭生计,我的哥哥在初中时也辍学外出打工,家里的经济才勉强好转一些。
但幸福并没有想象的那么简单。1999年的一天,哥哥突然胸口痛,爸爸让他赶紧去医院,哥哥一直没告诉我们他在医院的检查结果。一次,我给哥哥清洗枕套时,才看到哥哥藏起来的检查单,上面竟然写着:风湿性心脏病。
当时,我将这事告诉了爸爸,爸爸立马瘫坐在地,我们俩抱头痛哭,内心呐喊着:为什么老天要这么对我们一家人。
之后,我们把哥哥送去西昌市的一家大医院治病,医生告诉我们,哥哥的病很难治,要想治好只有换心脏,但是换心脏要二三十万。即使能筹到这个钱,也可能因为等不到合适的心脏,眼睁睁看着哥哥走。
我们拿不出那么多钱,只能先住院保守治疗。住院半个月没有好转,开了药回家养病,就这样拖了三年多。2004年正月初二,哥哥说心脏痛得撕心裂肺,送去医院急诊,医生说没救了,让我们回家。
2004年二月初四早上,哥哥走了,那时他才26岁。去世前的一个月,哥哥曾拉着我的手说:“你给我一包老鼠药吧,这样活着好累……”当时,我心如刀绞。

11年后,“不幸”又降临到了我的身上。
2015年的一天,我突然什么都看不到了,去医院诊断,我的左眼晶体全脱位,需要马上手术,否则将会失明。当时,西昌市做不了这个手术,我火速赶到了攀枝花的一家医院,医生看到我的外貌体征后,初步判断我可能是一名“马凡综合征”患者。
这是我人生中第一次听说“马凡综合征”。医生询问了我的家族史,还帮我做了相关检查,最终确诊我是一名罕见病病人——马凡综合征患者的事实。这是一种遗传性疾病,我的妈妈和哥哥也和我有着一样的特点:手长脚长,都长得很瘦、很高,我的妈妈身高1.7米,我的身高1.73米,我的哥哥1.8米,我的鞋子都比一般女性大出很多,43码。但我们怎么会知道,这竟可能是一种病?!
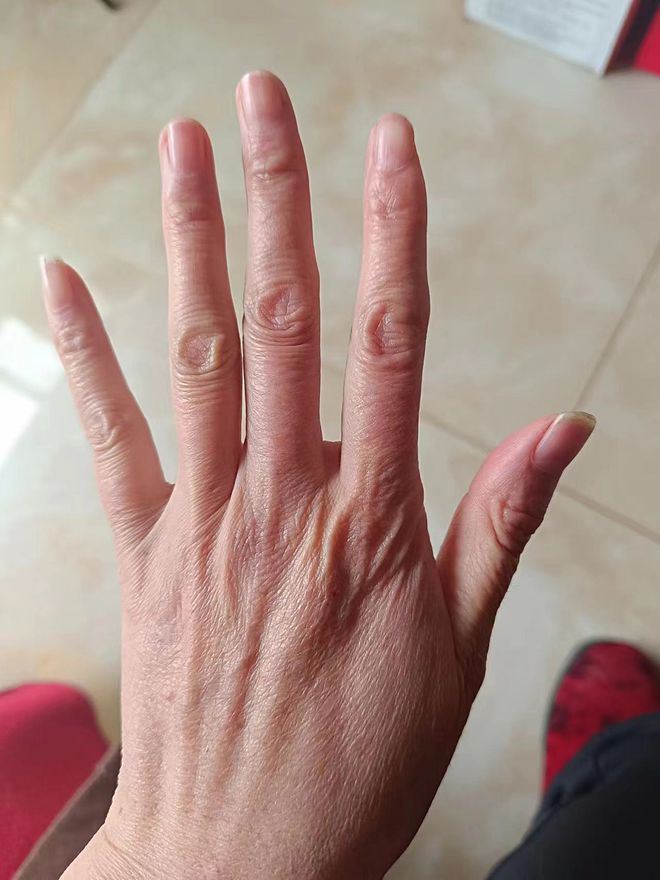
从小时候起,笑笑的手就比常人要大要长

笑笑的43码的鞋子
当时我的左眼视力0.01,右眼晶体半脱位。2015年和2016年我先后做了两次手术,治疗了我的双眼。
现在我想起来,是我小时候忽视了眼睛。从我记事起,我的视力就不好,黑板上的字一直看不清,大部分时候,我只能靠我的耳朵帮着去学习,或者问问同桌,黑板上的字一直靠我去蒙去猜,想到家里条件不好,从小我就一直瞒着家人这个事情。
在我第二次眼部手术期间,我的丈夫又被诊断出肺癌,跑了很多地方的大医院,前后治疗花了二十多万,家庭负债累累,但最终他还是离开了我和我们当时年幼的孩子。
历经两次心脏手术,继续吃药治疗
为了撑起这个家,我到广州打工,因为要尽快偿还债务,我选择了去快递公司上夜班,这个工作待遇稍微高一些,我不敢休息,上了两年多,没请过一天假,日子虽然很苦,但只要我和家人平安健康,我就很快乐了。

然而,我一度没有等来“好运”。2019年3月23日,我感觉到了腰痛,痛了一个礼拜,忍不了才去医院检查,还以为是骨质增生,吃点药就能好,但拿到检查报告时我傻眼了,医生告诉我,我患有主动脉瘤,需要尽快做手术。
我拿着报告单,安慰自己没事,可能检查出错了。过了几天还是腰痛,我就去了广州市中山医院再次检查,结果相同,医生告诉我情况很危急,需要准备30万做手术。
30万!这对我来说,简直是一个天文数字。我无法接受这样的事实,那时候非常想念自己的妈妈、丈夫,还有我的哥哥,但他们都离开了我。
孤独和无助伴随着我。我曾想过一了百了,但我还有孩子,孩子不能没有爸爸又没有妈妈。受限于现实条件,我打算放弃治疗,活到哪天算哪天。
随后有个很偶然的机会,一位病友建议我找北京康心马凡综合征关爱中心主任常继国寻求帮助,常主任同样也是一名马凡综合征患者。抱着试试的心情,我给他打了电话,当时我欠了很多外债,身上也没什么钱治病,常主任很热情地告诉我:“别怕,先去做一个超声看看,结果出来通知我。”
我还记得,当时因为我的儿子凌晨两点多骑车在路上晕倒、全身僵硬不能动弹,我忙着送孩子去医院、担忧孩子病情,把自己的事抛诸脑后。常主任还发来消息问我检查的进展,我告诉了他孩子的情况,说自己没有去检查。他耐心地告诉我,“手术也不是不能再等等,别怕,可以回家先养着,等半年复查看看结果,如果发生血管扩张就再进行手术。”他还说,会想办法帮我解决治疗费用的问题。
有一次,我在病友群里看到有人分享了全国马凡病友会的消息。这个会议每年都在上海德达医院召开,2019年8月,我第一次参加了这个病友会,当时我也想着能来亲眼看看常主任。通过这个大会,我学到了很多关于这个疾病的知识,大家一起交流病情,我的收获很大,不再害怕这个疾病,但说起手术,还是心有余而力不足。

诊断报告上明确了马凡综合征
2020年8月13日,我突发急性A型主动脉夹层。当时,我查阅了一些网上的资料:马凡综合征常伴有心血管系统异常,主动脉夹层就是其中一种致死的原因。运动、外伤、高血压和妊娠均会诱发主动脉夹层形成和急性主动脉破裂,导致患者因大出血休克而猝死,患者的自然寿命平均仅为32岁。
当时,我拿着身上仅剩下的2000元,坐火车,再转飞机,来到上海治疗。我还记得,我当时咨询了常主任,是否要做心脏手术,他的回答是要做,他让我去找上海德达医院的孙立忠院长,他专门做这方面的手术。
我到了医院急诊,那时我人是半清醒状态的,医生告诉我需要立刻手术。那时我儿子在老家筹钱,在我拿不出手术费的时候,孙立忠院长和他的团队就帮我做了手术,我很感恩他们对我的帮助。我的第二次胸腹主动脉置换术是在2022年8月4日完成的,很顺利。
历经两次心脏手术,我已经像正常人一样了。特别感谢常主任和孙院长,是他们让我重生。没有他们,我或许已经不在人世了!
尽管我知道,作为一名马凡综合征患者,我的外貌特征很难改变,如今经历了手术,我不再有腰痛的老毛病,也不用担心血管随时发生破裂。
现在,我和儿子相依为命,我们回到了四川老家,日子回归到了平淡。尽管我每天还需要吃药来进行治疗,但药物负担还在承受范围内,这一切我都很知足了!
医生告诉我,我的妈妈和哥哥离世,或许都与“马凡综合征”有关。如果有这一罕见病家族史、或者是存在胸廓脊柱畸形以及眼部病变如晶状体脱位、四肢长、手指脚趾长的人群,要及早诊断。
最后我希望,有更多的人看到我的故事,了解这种叫做马凡综合征的罕见病。如果能早日知道这种疾病的存在,我的两位至亲就不会早早离开。或许,人生还有很多可能。